

Material
- Vasos de Precipitados
- Matraz Kitasato
- Embudo Büchner
- Pipeta Graduada
- Probeta
- Termómetro
- Recipiente para baños
- Vidrio de reloj
- Frasco Lavador
Reactivos y disolventes
- CoCl₂ ∙ 6H₂O
- Ácido Clorhídrico concentrado
- NH₄Cl
- NH₃ concentrado
- H₂O₂
- NaNO₂
Precauciones: Todas las reacciones en las que intervengan gases tóxicos, deben de realizarse en vitrina.
| CoCl₂ ∙ 6H₂O | Nocivo por ingestión.No se debe de respirar el polvo.Posible agente citotóxico (mutagénico, teratógeno y carcinógeno). |
| HCl | Evitar respirar el vapor y el contacto con la piel y los ojos. Irrita el sistema respiratorio y produce graves quemaduras.Es tóxico por inhalación. |
| NH₃ | Evitar respirar el vapor y el contacto con la piel y los ojos.Irrita el sistema respiratorio. |
| NH₄Cl | Nocivo por ingestión.No respirar el polvo.Irrita los ojos. |
| H₂O₂ | Evite el contacto con la piel y los ojos.Produce quemaduras. |
| NaNO₂ | Es tóxico por inhalación.Puede arder en presencia de material combustible.Posible teratógeno. |
En este experimento de laboratorio sintetizamos 4 tipos diferentes de complejos de cobalto(III), sus estructuras vienen representadas a continuación, nota que cada complejo es precursor del anterior:

En esta práctica experimental seremos capaces de estudiar el efecto del ligando en las propiedades espectroscópicas de los complejos en la franja del UV-VIS.
Dada su naturaleza oxidante, el ion Co³⁺ es capaz de oxidar al agua en el proceso redox de la figura, el cual está favorecido termodinámicamente. Es muy inestable en agua, por eso se añade amoniaco durante el experimento para estabilizar y que no se reduzca.

El cobalto(III) es un metal d⁶, con disposición octaédrica. Ésto nos brinda dos posibles disposiciones; alto spin o bajo spin.
La experiencia nos muestra que todos los complejos de Co(III) son de bajo spin y diamagnéticos, dado su alto estado de oxidación (indica campo fuerte).
Se comprobará mediante medidas espectroscópicas en la franja del (VIS-UV) que realmente se confirma la disposición electrónica de bajo spin.
Además, mediante I.R., se podrá diferenciar el grupo nitrito del nitro en los complejos (3) y (4).
También se realizarán medidas de conductividad y se comparan con los resultados teóricos para electrolitos (3:1) en el caso de (1) o electrolitos (2:1) para (2), (3) y (4).
Como se ha visto, el ion Co(III) es bastante inestable. Por ello, para estabilizar el catión, se utilizan ligandos dadores (nitrógeno dadores en este caso) como el amoniaco.

La idea general de las síntesis es ir coordinando ligandos de amoníaco al Co(II) y después, oxidar de Co(II) a Co(III). Lo primero que se hace es partir de una sal de cobalto(II) soluble en agua, después añadir ligando amoniaco, estabilizar y finalmente oxidar con agua oxigenada.
Síntesis de [Co(H₂O)(NH₃)₅]Cl₃ (1)
Para llevar a cabo la síntesis del primer complejo, es necesario disolver en 15 mL de agua destilada los 3.0 gramos de CoCl₂ ∙ 6H₂O (color violeta). Todo dentro de un frasco lavador.

Una vez disuelto, se añaden 1.8 g de NH₄Cl y 15 mL de NH₃ concentrado. Durante la adición, agitar.

- La última molécula de amoníaco desprotona la última de agua. El agua al coordinarse al metal tiene más acidez, siendo más fácil de desprotonar.
- Se usa una disolución reguladora NH₃/NH₄⁺ para evitar la formación de [Co(NH₃)₄(OH)₂] a pH básicos
Tras esto, se añaden, gota a gota, 6 mL de H₂O₂ al 30%. (Con oxígeno sería más lento)

Cuyo proceso redox (sin ajustar) es:

Es importante NO CALENTAR esta reacción para no obtener erróneamente el complejo (2).
De esta forma, la concentración de aniones hidróxido va en aumento.
Teniendo presente que en el medio de reacción hay:

Además, esta reacción está favorecida ya que es exotérmica, es decir, emite calor cuando tiene lugar.
Al cesar la efervescencia, la oxidación ha finalizado y es entonces cuando se debe borbotear una corriente de aire a través de la disolución, durante 45 minutos. De esta forma se elimina el amoníaco en exceso para que no se forme demasiado cloruro de amonio. Este cristalizaría y no se podría obtener el complejo por efecto del ión común.
Una vez eliminado, se enfría la disolución en baño de hielo y se neutraliza con HCl concentrado. El estado de oxidación +3 es estable a pH neutro.
Se continúan añadiendo 14 mL de HCl concentrado hasta que precipita un sólido rojo-naranja que corresponde a nuestro compuesto (1) cloruro de acuopentamincobalto(III).
Éste exceso de HCl hace que, debido al efecto del ión común, se desplace el equilibrio de la reacción hacia la formación del precipitado.

El sólido se filtra a vacío, se lava con una disolución bien fría (para que no se añadan más cloruros) de HCl 6M (2x10mL), isopropanol (2x10mL) y finalmente se seca al aire en trompa de agua. No se puede usar agua para lavar ya que disuelve al compuesto.
Se obtuvieron 2.35 g del compuesto (1).
Síntesis de [CoCl(NH₃)₅]Cl₂ (2)
Para realizar la síntesis del complejo número (2), se parte del complejo (1).
Se coloca el compuesto (1) en un vidrio de reloj y se calienta en estufa durante 1 noche a 110ºC.
Tras esto, se obtuvieron 2.18 g de [CoCl(NH₃)₅]Cl₂, un compuesto sólido de aspecto violeta.

De esta forma se sustituyó un cloruro, si continuamos calentando por encima de 110 º C se sustituyen dos.
El enlace del amoniaco es más fuerte que con el agua. El cobalto prefiere estar coordinado a ligandos aniónicos. Cuanto más se caliente más átomos de cloro se unirán perdiendo amoniaco. Si se calienta mucho se obtiene el cloruro correspondiente.

Síntesis de [Co(κO-NO₂)(NH₃)₅]Cl₂ (3)
Este compuesto, es un producto cinético y por lo tanto es inestable y conlleva la formación, al cabo de un tiempo, del producto termodinámico (4).
Se tiene el complejo (2), primero se forma el acuocomplejo (1º añadir OH⁻ hidrolizando al amoniaco y 2º añadir protones con HCl) y después se añade el NO₂⁻.
Para empezar, se colocan siguiendo el siguiente orden; 0.5g del complejo (2), 30 mL de agua destilada y 3 mL de amoníaco concentrado.
Tras la adición, se calienta hasta disolución del sólido, dándose las siguientes reacciones:


El esquema general es el siguiente:

Al añadir amoniaco concentrado sobre una disolución acuosa, se producen iones hidróxido que sustituyen al cloro del complejo (2).
La disolución resultante se enfría a 10 ºC y se añade con cuidado HCl 6M hasta neutralización (Neutraliza y protona).

Una vez neutralizada la disolución, se añade 1 g de nitrito de sodio, 2 mL de HCl concentrado y la mezcla se deja en baño de hielo durante 30 min, formándose un precipitado color rojo salmón.

Nota cómo en el primer paso se sustituye un ligando aniónico por uno neutro, lo cual está favorecido.
El sólido obtenido se lava con isopropanol (2x5mL) y se seca en trompa de aire.
Se obtuvieron 0.90 g del compuesto [Co(κO-NO₂)(NH₃)₅]Cl₂.
Síntesis de [Co(κN-NO₂)(NH₃)₅]Cl₂ (4)
Se mezclan 0.5 g del complejo (3) con 10 mL de agua destilada. Para formar el producto termodinámico tengo que calentar y así se favorece la isomerización.

La mezcla se calienta, SIN LLEGAR A HERVIR, durante 20 minutos.
Tras esto, se deja enfriar la disolución, se añaden 5 mL de HCl concentrado y se deja en baño de hielo.
Al enfriarse la mezcla precipita un sólido de color amarillo amarronado.

Los cristales obtenidos se filtran, se lavan con isopropanol (2x5mL) y se secan al aire en trompa de agua.
Tras esta síntesis será necesario verificar mediante técnicas de I.R. si la reacción se ha completado totalmente.
Si quedaran restos del compuesto de partida, y observamos las bandas del precursor nitro, se debe de introducir el compuesto en estufa durante 1 hora a 100ºC.
Finalmente, se obtuvieron 0.33 g del complejo (4).
Caracterización
Los productos sintetizados se caracterizaron mediante medidas de conductividad eléctrica molar preparando disoluciones acuosas de los complejos de concentraciones del orden de 1×10⁻³ M.
Además las reacciones y los compuestos sintetizados se siguieron mediante espectroscopía infrarroja con pastillas de KBr y VIS-UV (en agua).
Espectroscopía Infrarroja
Al pasar del complejo (1) al (2), lo que hacemos es sustituir un ligando agua por un cloro, éste último es de campo débil y hace aparecer una señal en el espectro número 2 que no estaba en el espectro número 1 a 2356 cm⁻¹, y en general, todas las señales que ya había anteriormente van hacia longitudes mayores (en cm⁻¹), lo que indica que el enlace se debilita.
Δ₀↑ ⇒ E(enlace)↓ ⇒ λ↑


En el espectro I.R. número 3, aparecen dos picos característicos del grupo nitrito, uno a 1065 cm⁻¹ y otro por debajo de 1450 cm⁻¹.
La señal de 1065 cm⁻¹ debe desaparecer en el espectro nº 4 si queremos confirmar la total transformación del compuesto (3) nitrito al (4) nitro.
Finalmente se verifica en el espectro nº 4, que la reacción finalizó. Se observa una clara aparición de pico a unos 822 cm⁻¹.
El otro pico característico es más difícil de distinguir, ya que se asemeja mucho en longitud, pero puede verse cómo se ha movido de 1449 cm⁻¹ en el espectro nº 3 a 1427 cm⁻¹ en el espectro nº4.

Espectro I.R. nº1 del complejo (1) sintetizado en el primer paso.

Espectro I.R. nº 2 del complejo (2) sintetizado en el segundo paso.

Espectro I.R. nº 3 del complejo (3) sintetizado en el tercer paso, es este caso aparece el ligando nitrito. (arriba)
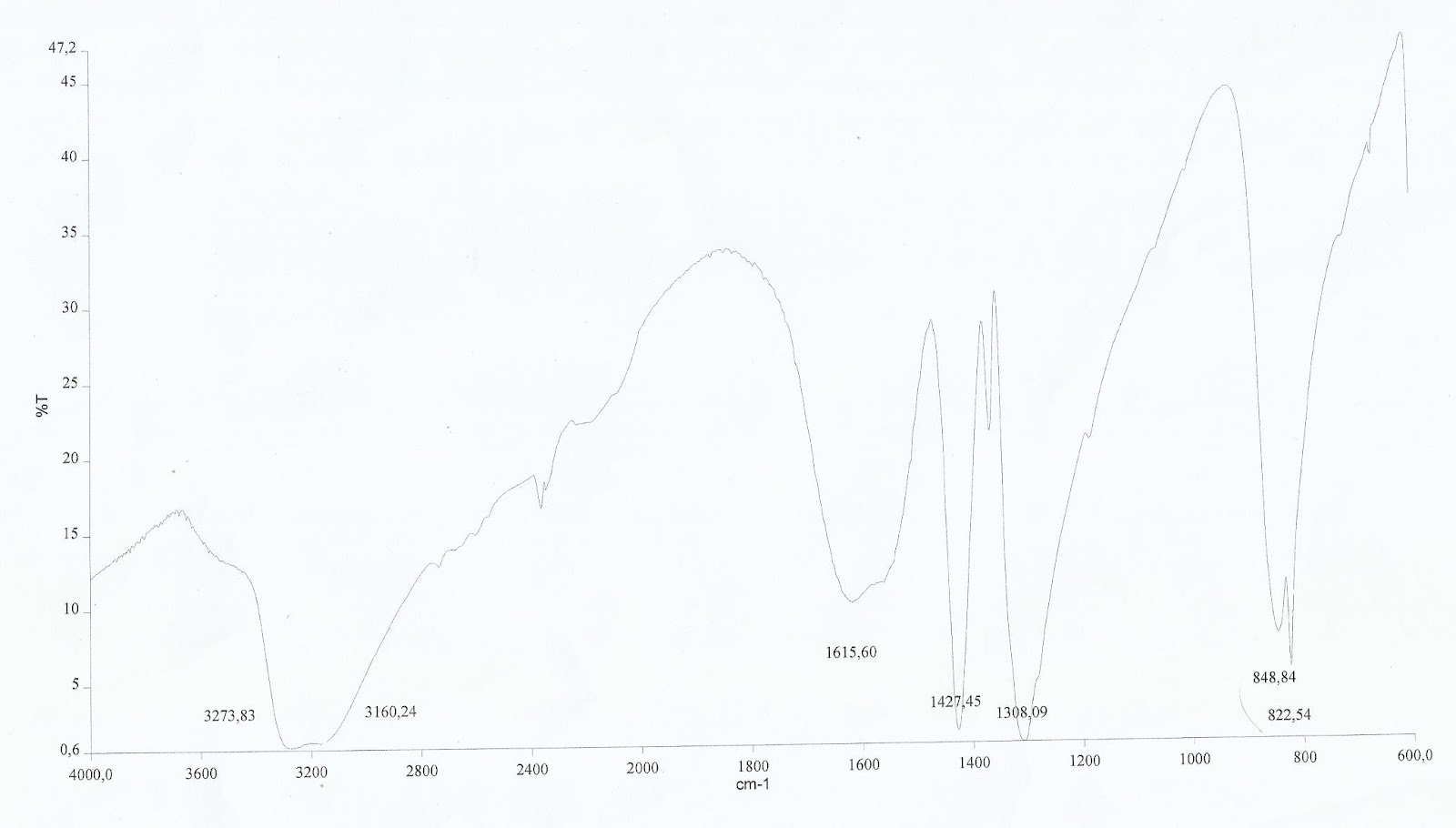
Espectro I.R. nº 4 del complejo (4) sintetizado en el último paso, en él se observa la aparición de la banda característica del grupo nitro.
Espectroscopía VIS-UV
Los espectros están ordenados por números de acuerdo a los complejos de igual forma que los infrarrojos, por lo que el compuesto 1 corresponde al espectro 1 y así sucesivamente. Al fijarnos en los espectros VIS-UV vemos cómo varía el desdoblamiento octaédrico al ir sustituyendo el ligando Cl/H₂O/ONO/NO₂. Si el desdoblamiento baja, la energía también y por lo tanto la longitud de onda aumenta.
El compuesto (1) de color rojo, muestra dos señales: una a 345 nm que corresponde a la transición ¹A₁g ⇒ ¹T₂g y otra a 492 nm para la transición ¹A₁g ⇒ ¹T₁g.
El compuesto (2) es de color violeta, muestra dos señales: una a 364 nm para la transición ¹A₁g ⇒ ¹T₂g y otra a 280 nm para Cl/Co⇒ Co³⁺.
El compuesto (3) es de color rojo salmón, y en el espectro se puede apreciar una señal característica a 493 nm que corresponde a la transición ¹A₁g ⇒ ¹T₁g.
Finalmente, el compuesto (4) es de color marrón, en el espectro se aprecia una banda débil a 451 nm resultado de la transición ¹A₁g ⇒ ¹T₁g.

Espectro nº 1

Espectro nº 2

Espectro nº 3

Espectro nº 4
Medidas de conductividad
Se tomaron 0.0062 g de cada complejo y se disolvieron en un matraz aforado de 25 mL para llegar a la concentración deseada que deberá ser del orden de 1 x 10⁻³ M. Para la medida de la conductividad, se toma la medida del disolvente puro, en nuestro caso agua destilada y se le resta a la medida que dé la disolución acuosa del complejo.
Ésta medida experimental debe corresponder con los valores tabulados para electrolitos, clasificados en función de si son electrolitos (1:1), (2:1), (3:1)… etc
| Complejo | Λ(dte) (μS/cm) | Λ(complejo) | Λ(experimental) | Λ(teórica) |
| 1 | ||||
| 2 | 228 | 301 | 73 | 70 |
| 3 | 200 | 328 | 128 | 120 |
| 4 | 216 | 217 | 1 | 0 |
Bibliografía
- Experimental Methods in Inorganic Chemistry, J. Tanaka, S. L. Suib, Ed. Prentice Hall, 1999.
- Inorganic Experiments, 3rd ed. Ed. J. Derek Woolins, Wiley-VCH, 2010.
- G. M. Williams, J. Olmsted III, A. P. Breksa III, J. Chem. Ed. 1989, 66, 1043.
¿Cómo extraer la cafeína del café?

El Fascinante Mundo del Ciclo del Cobre: Explorando la Química Inorgánica

Bombas Caseras: Experimentos con Precaución

Las 7 reacciones químicas más espectaculares que parecen magia

¿Qué es una pila de limón?

Síntesis de los complejos [Co(H₂O)(NH₃)₅]Cl₃, [CoCl(NH₃)₅]Cl₂, [Co(κO-NO₂)(NH₃)₅]Cl₂ y [Co(κN-NO₂)(NH₃)₅]Cl₂.

Síntesis del complejo iluro [Ag{CH(PPh₃)C(O)CH₃}₂][NO₃]. Caracterización mediante medidas de conductividad eléctrica en disolución y sus espectros de IR y de RMN (¹H y ³¹P{¹H}).

Síntesis del complejo dinuclear [Cr₂(μ-O₂CCH₃)₄(H₂O)₂] y determinación de su susceptibilidad magnética.

Preparación de [Fe(ղ⁵-C₅H₅)(ղ⁵-C₅H₄COCH₃)] a partir de ferroceno. Purificación por cromatografía de columna y caracterización mediante IR y RMN

Síntesis del óxido doble CaMnO₃

Síntesis de oxalatos hidratados de iones metálicos del grupo 2 (MC₂O₄ . nH₂O; M = Ca, Sr, Ba). Caracterización mediante análisis termogravimétrico.
Buscando el Xenón desaparecido en el Manto Terrestre: Estudio Termodinámico
Complejos de manganeso activos en la reducción electrocatalítica de CO₂

Síntesis de dos complejos de níquel(II) con estructuras diferentes: [Ni(NCS)₂(PPh₃)₂] y [NiCl₂(PPh₃)₂]. Caracterización mediante medidas de susceptibilidad magnética y espectroscopía IR.

Síntesis de los complejos fac-[MnBr(CO)₃(κ²P,P-dppm)] y cis,mer-[MnBr(CO)₂(κ²P,P-dppm){P(OPh)₃}].
¿ Qué Materiales hay en un Laboratorio Químico?

Obtención de Alumbre Amónico y Azul de Thènard

Obtención de Cloruro de Cobre (I) CuCl.

Obtención de Ditionito de Zinc

Obtención de Sales de Plomo
![síntesis de (NH4)2[SnCl6]](https://www.quimiclan.com/wp-content/uploads/2020/02/síntesis-de-NH42SnCl6-390x200.jpg)
Obtención de (NH₄)₂SnCl₆

Obtención de Sales de Cobre (II)

Obtención de Gel de Sílice

Obtención de Ácido Bórico

Obtención de Sodio Amoniacal

Obtención de Tiosulfato de Sodio Pentahidratado

Obtención de Oxalato Férrico de Potasio Trihidratado

